Epigenética del cáncer
Manel Esteller

INTRODUCCIÓN
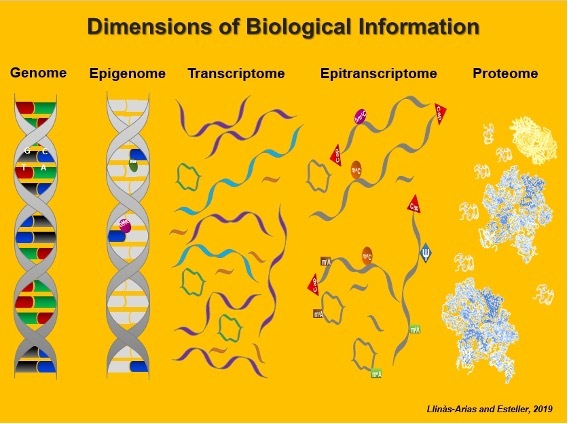
El grupo continúa el exhaustivo estudio de la epigenética que Manel Esteller ha llevado a cabo como investigador principal durante toda su carrera científica. En la actualidad, las investigaciones se centran en el establecimiento de mapas epigenómicos y epitranscriptómicos para células normales y transformadas, en el estudio de las interacciones entre las modificaciones epigenéticas y los ARN no codificantes y en el desarrollo de nuevos fármacos epigenéticos para el tratamiento del cáncer.
NUESTRA INVESTIGACIÓN
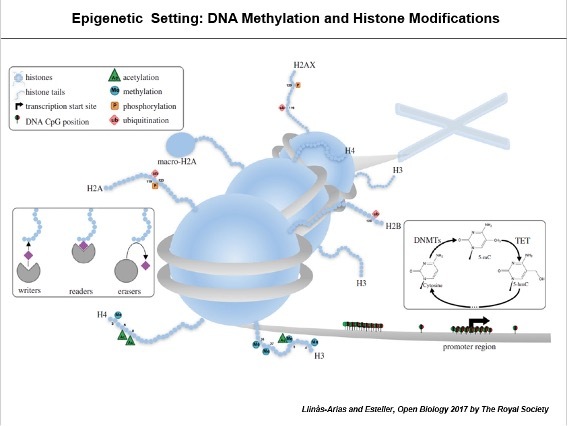
Nuestro laboratorio es uno de los responsables de establecer que la interrupción epigenética de la transcripción del ARNm, en particular los patrones de metilación del ADN y de modificación de histonas, contribuye al inicio y la progresión de los tumores en humanos (revisado en Esteller, N Engl J Med 2008; Heyn y Esteller, Nat Rev Genet 2012; Berdasco y Esteller, Nat Rev Genet 2019); Davalos y Esteller, CA Cancer J Clin 2023).
También se ha identificado que los micro-ARN (ARN pequeños no codificantes que regulan la expresión génica mediante emparejamiento de bases específico de secuencias en dianas de ARNm) desempeñan asimismo un papel clave en la biología celular y pueden afectar al desarrollo del cáncer. En este contexto, llevamos a cabo la caracterización del primer micro-ARN sometido a silenciamiento asociado a metilación específico del cáncer (Lujambio et al., Cancer Res 2007), seguido de la caracterización de otros muchos micro-ARN alterados de forma similar (Lujambio et al., PNAS 2008; Davalos et al., Oncogene 2012).
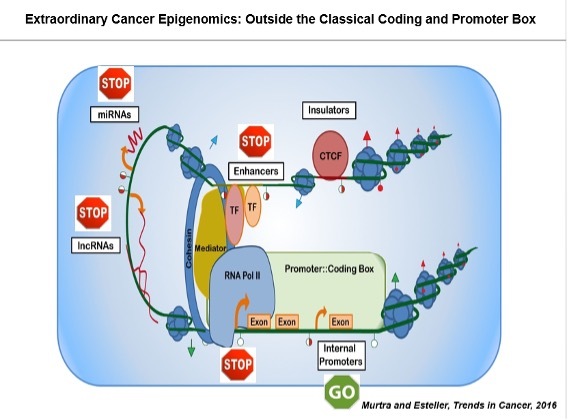
También hemos estudiado otros tipos de ARNnc, tales como diversas subclases de ARN no codificante de cadena larga (ARNncl), sometidos a eventos de metilación aberrante del ADN en el cáncer en humanos (Lujambio et al., Oncogene 2010; Guil et al., Nat Struc Mol Biol 2012; Liz et al., Mol Cell 2014; Diaz-Lagares et al., PNAS 2016). Hemos demostrado que, en ocasiones, estas lesiones epigenéticas tienen lugar fuera de los promotores mínimos y se producen en potenciadores (Heyn et al., Genome Biol 2016; Vidal et al., Oncogene 2017) o en promotores internos crípticos (Vizoso et al., Nature Medicine 2015).
NUESTROS OBJETIVOS
Nuestro grupo está interesado desde hace tiempo en trasladar el uso del conocimiento de la epigenética logrado gracias a la investigación al uso de biomarcadores para predecir desenlaces clínicos y para evaluar nuevos fármacos que reviertan el distorsionado panorama epigenético (Berdasco y Esteller, Nature Review Genetics 2019). Así, por ejemplo, hemos usado marcadores epigenéticos para predecir la respuesta a terapias antitumorales tras la observación inicial de que la metilación del gen MGMT predecía la respuesta a agentes alquilantes en el glioma (Esteller et al., N Engl J Med 2000).
Hemos demostrado la relación entre la metilación de MGMT y la respuesta a agentes alquilantes en el linfoma (Esteller et al., J Natl Cancer Inst, 2002), entre la metilación de WRN y la respuesta al irinotecán (Agrelo et al., Proc Natl Acad Sci USA, 2006), entre la metilación de BRCA1 y la respuesta a inhibidores de PARP (Veeck et al., J Natl Cancer Institute, 2010) y entre la metilación de DERL3 y la respuesta a inhibidores de la glucólisis (Lopez-Serra et al., Nature Communications, 2014). También hemos identificado la metilación de SRBC (Moutinho et al., J Natl Cancer Institute, 2014) y de SLFN11 (Nogales et al., Oncotarget 2015) como marcadores de resistencia para derivados de platino en tumores en humanos y hemos identificado el regulador de EGFR TBC1D16 como un sensibilizante para terapias con inhibidores de BRAF y MEK (Vizoso et al., Nature Medicine 2015). La pérdida epigenética de SVIP también se ha relacionado con la respuesta a inhibidores de GLUT1 (Llinas-Arias et al., JCI Insight 2019). Desde el punto de vista de la multiómica, hemos contribuido a la caracterización de la sensibilidad farmacológica en 1000 líneas celulares de cáncer (Iorio et al., Cell 2016) y hemos averiguado por qué algunos pacientes responden de manera excepcional al tratamiento (Wheeler et al., Cancer Cell 2021) y descifrado los sustratos moleculares que subyacen en los dobles o humanos extremadamente similares (Joshi et al., Cell Reports 2022), y caracterizado a nivel multiómico single-cell el efecto de un agente hipometilante en pacientes con síndrome mielodisplásico (Campillo-Marcos et al., Cancer Research Communications 2024).
NUESTROS RETOS
Continuando con este aspecto traslacional de nuestro trabajo, también estamos interesados en el desarrollo y el estudio de nuevos fármacos epigenéticos dirigidos a escritores, lectores y borradores de la modificación de histonas y la metilación de ADN, y que podrían tener un efecto antineoplásico (Lara et al., Oncogene 2008; Zubia et al., Oncogene 2009; Huertas et al., Oncogene 2012; Perez-Salvia et al., Oncotarget 2017; Perez-Salvia et al., Haematologica 2018).
Resulta interesante que el «repertorio» de modificaciones epigenéticas del ADN sea bastante limitado, tal como pusimos de manifiesto en una revisión reciente (Heyn y Esteller, Cell 2015). Esto contrasta drásticamente con las más de cien modificaciones post-transcripcionales que se producen en el ARN (Esteller y Pandolfi, Cancer Discovery 2017; Davalos et al., Cell 2018; Rosselló-Tortella, Ferrer y Esteller, Blood Cancer Discovery 2020), Orsolic et al., Trends in Genetics, 2023).
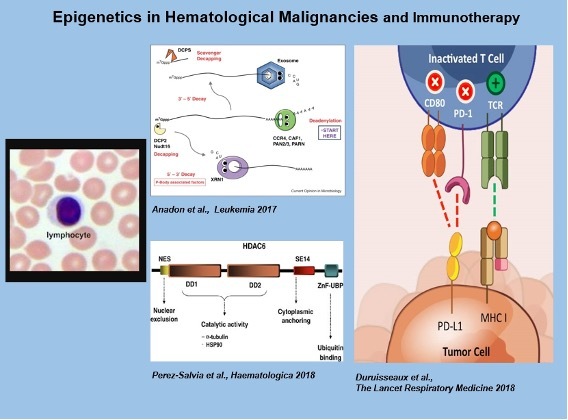
Hasta hace muy poco tiempo, resultaba casi imposible crear un mapa adecuado de las modificaciones epigenéticas de la molécula de ARN, algo que suponía un obstáculo para numerosos estudios en esta área e impedía lograr avances en el estudio de la importancia de cada una de dichas modificaciones del ARN. No obstante, las metodologías recientes nos permiten actualmente el estudio del llamado «epitranscriptoma». En este campo, hemos identificado la edición aberrante del ARN mediada por amplificación de ADAR1 en el cáncer de pulmón (Anadon et al., Oncogene 2016), el decapping de ARN alterado mediado por silenciamiento epigenético por NUDT16 en la leucemia linfocítica aguda de linfocitos T (LLA-T) (Anadon et al., Leukemia 2017), la pérdida de metilación de ARN en ARN ribosómico en el glioma (Janin et al., Acta Neuropathol 2019), la modificación de guanina sin emparejamiento de ARN de transferencia en el cáncer de colon (Rosselló-Tortella et al., PNAS 2020), los defectos m1A en el linfoma de Hodgkin (Esteve-Puig et al., Blood 2020), la desregulación epigenética de los ARNt en diversos tipos de tumores (Rosselló-Tortella et al., Molecular Cancer 2022), el silenciamiento de la 5mC ARN metiltransferasa NSUN7 en cáncer de hígado (Portiz-Barahona et al., Molecular Cancer 2023) y la contribución de las desviaciones de ARN m6A en la transdiferenciación celular (Bueno-Costa et al., Leukemia 2022). Los conocimientos en esta área son limitados y su estudio es objeto de una profunda investigación en nuestro laboratorio.
También estamos interesados desde hace tiempo en investigar trastornos monogénicos que afectan a genes epigenéticos (Urdinguio et al., Lancet Neurol. 2009), en particular, en el síndrome de Rett. Esta enfermedad se asocia a una mutación de línea germinal en MECP2, una proteína que es atraída hacia el ADN metilado. A lo largo de los años, hemos identificado las dianas génicas de la MECP2 (Ballestar et al., EMBO J 2013; Petazzi et al., RNA Biol. 2013, Neurobiol Dis. 2014), hemos estudiado en detalle la genómica del síndrome de Rett (Saez et al., Genet Med 2016; Lucariello et al., Hum Genet 2016) y hemos llevado a cabo estudios preclínicos de fármacos (Szczesna et al., Neuropsychopharmacology 2014; Jorge-Torres et al., Cell Reports 2018).
En un contexto similar, también sentimos curiosidad por los perfiles epigenómicos de enfermedades comunes como las alteraciones cardiovasculares (Zaina et al., Circ Cardiovasc Genet. 2014; Valencia-Morales et al., BMC Med Genomics 2015) y el Alzheimer y otras enfermedades neurodegenerativas (Sanchez-Mut et al., Brain 2013; Hipoccampus 2014; Transl. Psychiatry 2016; Nature Medicine 2018).
Finalmente, estamos muy interesados en establecer nuevas plataformas epigenómicas para elaborar mapas detallados del metiloma del ADN. De hecho, nuestro laboratorio es pionero en la validación de los microarrays de metilación del ADN empleados habitualmente, tales como el microarray 450K (Sandoval et al., Epigenetics 2011) y el microarray EPIC/850K (Moran et al., Epigenomics 2016) y su nueva versión V2 (Noguera-Castells et al. Epigenetics, 2023), además del microarray de metilación de ADN de ratón (García-Prieto et al., Epigenetics 2022). El uso de estos enfoques ha hecho posible lograr diversos avances científicos, tales como: el establecimiento de firmas de metilación del ADN que están asociadas a la diseminación temprana en el cáncer de pulmón (Sandoval et al., JCO 2010); el diagnóstico del tipo de tumor en el cáncer de origen primario desconocido (Moran et al., Lancet Oncology 2016); una mejor comprensión de la respuesta a la inmunoterapia anti-PD1 (Duruisseaux et al., The Lancet Respiratory Medicine 2018); la obtención del primer metiloma del ADN de células CAR-T con valor clínico (Garcia-Prieto et al., J National Cancer Institute 2021), la predicción de la gravedad clínica de la COVID-19 en base a la epigenética en adultos (Castro de Moura et al., Lancet EBioMedicine 2021) y niños (Davalos et al., Lancet EclinicalMedicine 2022) o las cicatrices epigenéticas en el pulmón por la infección SARS-CoV-2 (Noguera-Castells et al., CHEST 2023).
Equipo

Manel Esteller
Jefe de grupo senior
Jefe de grupo senior

Esteban Fernando Setién
Investigador/a Asociado/da
Investigador/a Asociado/da

Eva Musulén
Investigador/a Asociado/da
Investigador/a Asociado/da

Gerardo Ferrer
Investigador/a Asociado/da
Investigador/a Asociado/da

Maria Verónica Dávalos
Investigador/a Asociado/da
Investigador/a Asociado/da

Aleix Noguera
Investigador/a Postdoctoral
Investigador/a Postdoctoral

Alex Martinez-Sabadell
Investigador/a Postdoctoral
Investigador/a Postdoctoral

Enrique Javier Arenas
Investigador/a Postdoctoral
Investigador/a Postdoctoral

Eva Crespo
Investigador/a Postdoctoral
Investigador/a Postdoctoral

Ignacio Campillo
Investigador/a Postdoctoral
Investigador/a Postdoctoral

Ines Oršolić
Investigador/a Postdoctoral
Investigador/a Postdoctoral

Reid Elizabeth Blanchett
Investigador/a Postdoctoral
Investigador/a Postdoctoral

Aleix Franquesa
Investigador/a predoctoral
Investigador/a predoctoral

Carlos Quero
Investigador/a predoctoral
Investigador/a predoctoral

Eloy Santos
Investigador/a predoctoral
Investigador/a predoctoral
Karim Yassine
Investigador/a predoctoral
Investigador/a predoctoral

Lovro Martinovic
Investigador/a predoctoral
Investigador/a predoctoral
Manal Agdada
Investigador/a predoctoral
Investigador/a predoctoral

Tamar Levy
Investigador/a predoctoral
Investigador/a predoctoral

Victor Sanchez
Investigador/a predoctoral
Investigador/a predoctoral

Yoana Veselinova
Investigador/a predoctoral
Investigador/a predoctoral

Marta Soler
Técnico/a de laboratorio superior
Técnico/a de laboratorio superior
Publicaciones seleccionadas
Ayudas vigentes
CPP2023-010525
Ministerio de ciencia e innovación
COMBATIR NUEVAS ESTRATEGIAS COMBINATORIAS Y BIOMARCADORES DE RESPUESTA A INHIBIDORES DE HDAC6 PARA EL TRATAMIENTO DEL CÁNCER.
European academy of dermatology and venereology
SCIENCE-SS Single-cell Characterization of Epigenetic and Transcriptomic Dynamics in Sézary Syndrome Progression
HR24-00788
Fundació "la caixa"
Solve-ALD Brain Inflammation in adrenoleukodystrophy: from drivers to treatments
PMPER24/00017
Instituto de salud carlos iii
SEED-ALS Synergizing Efforts to Develop and Accelerate Breakthroughs in ALS Research
GLD24/00098
Gilead sciences s.l.
Identification of DLBCL key molecular features for CAR-T cell therapy
101136622
European commission
THRIVE TUMOUR-HOST INTERACTIONS IN LIVER CANCER OF CHILDHOOD AND ADULTS
HR24-00788
Fundació "la caixa"
Solve-ALD Brain Inflammation in adrenoleukodystrophy: from drivers to treatments
A29372/GEACC19003CED
Fundación científica de la asociación española contra el cáncer
PREDICT-Meso PRE-malignant Drivers Combined with Target-Drug validation in Mesothelioma
OC-2023-1-26592
European commission
SENESCENCE2030 Targeting Cell Senescence to Prevent Age-Related Diseases
DJCLS 03 R/2024
Deutsche josé carreras leukämie stiftung
scEpiCART Refinement of Epigenetic Factors to Improve CAR T Cell Therapeutic Efficacy
AP183262023
Fundación mutua madrileña
Identificación de determinantes de respuesta a quimioinmunoterapia en biopsia líquida, en pacientes con cáncer microcítico de pulmón.
RETOS245779LLOV
Ministerio de ciencia e innovación
ASPIRE Neoadjuvant-adjuvant immunotherapy and cancer vaccines to improve survival in hepatocellular carcinoma
CB16/12/00312
Instituto de salud carlos iii
CIBERONC Centro de Investigación Biomédica en Red Cáncer
Cll global research foundation
Gene Expression Regulation Study Mediated by the RNA-Binding Protein Musashi2 and the RNA Methyladenosine Transferase 3, as Therapeutic Targets in Chronic Lymphocytic Leukemia
HR22-00732
Fundació "la caixa"
MyoClonal Somatic mutations and clonal hematopoiesis as predictors and drivers of heart failure progression
FJC2020-044658-I
Ministerio de ciencia e innovación
Single cell analysis of clonal heterogeneity in myelodysplastic syndromes treated with azacitidine
101126676
European commission
IRB-TARGET IRB Barcelona International PhD programme: on TARGET for high-impact biomedicine
TALEN258406BLAN
Fundación científica de la asociación española contra el cáncer
T-REX Transcriptional Role and Epigenetics of the X chromosome in leukemias
2025 STEP 00422
Agència de gestió d'ajuts universitaris i de recerca
Methylation Maps for Precision Hematology: Decoding Epigenetic Signatures of MDS Progression and Venetoclax Resistance in AML
2024 FI-1 00954
Agència de gestió d'ajuts universitaris i de recerca
Overcoming Cancer Immunotherapy Resistance
PRE2022-105015
Ministerio de ciencia e innovación
USO DE APROXIMACIONES DE CELULA UNICA PARA DESCIFRAR LA EPIGENOMICA DEL CANCER Y LAS EPIDROGAS
FPU22/01655
Ministerio de universidades
Estudios de epigenética del cáncer a nivel de célula única