Cancer epigenetics
Manel Esteller

INTRODUCTION
The group continues the wide-ranging work on epigenetics that Manel Esteller, the group leader, has carried out during his career until now. Current research is devoted to the establishment of the epigenome and epitranscriptome maps for normal and transformed cells, the study of the interactions between epigenetic modifications and non-coding RNAs, and the development of new epigenetic drugs for cancer therapy.
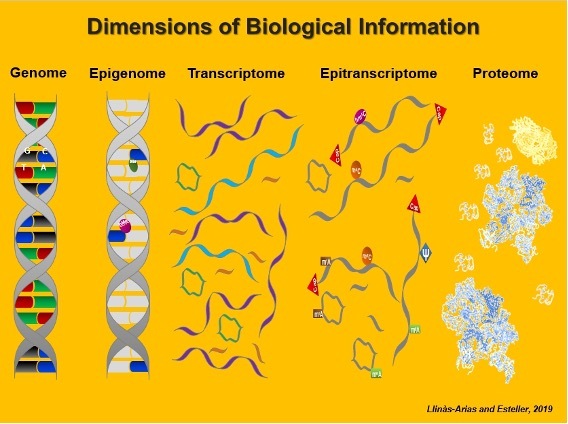
OUR RESEARCH
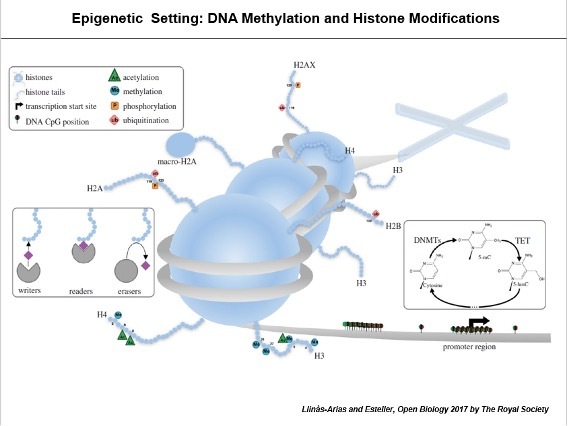
Our laboratory is one of those responsible for establishing the observation that epigenetic disruption of mRNA transcription, particularly in DNA methylation and histone modification patterns, contribute to the initiation and progression of human tumours (reviewed in Esteller, N Engl J Med 2008; Heyn and Esteller, Nat Rev Genet 2012; Berdasco and Esteller, Nat Rev Genet 2019; Davalos and Esteller, CA Cancer J Clin 2023).
It has also been recognized that microRNAs (small non-coding RNAs that regulate gene expression by sequence-specific base pairing in mRNA targets) also play a key role in the biology of the cell, and can have an impact on the development of cancer. In this context, we characterized the first miRNA undergoing specific cancer-methylation associated silencing (Lujambio et al., Cancer Res 2007), followed by the characterization of many other miRNAs disrupted in the same manner (Lujambio et al., PNAS 2008; Davalos et al., Oncogene 2012).
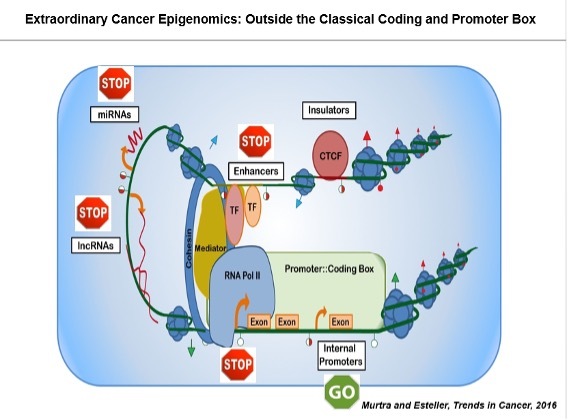
We have also studied other types of ncRNA, such as subclasses of lncRNA, undergoing aberrant DNA methylation events in human cancer (Lujambio et al., Oncogene 2010; Guil et al, Nat Struc Mol Biol 2012; Liz et al., Mol Cell 2014; Diaz-Lagares et al., PNAS 2016). We have shown that sometimes these epigenetic lesions occur outside the minimal promoters and take place in enhancers (Heyn et al., Genome Biol 2016; Vidal et al, Oncogene 2017) or at cryptic internal promoters (Vizoso et al., Nature Medicine 2015).
OUR GOALS
Our group has had a long-standing interest in translating the use of epigenetic knowledge gained from research into biomarkers to predict clinical outcome and to assay new drugs to reverse the distorted epigenetic landscape (Berdasco and Esteller, Nature Review Genetics 2019). For example, we have used epigenetic markers to predict response to anti-tumour therapies and following the initial observation that MGMT gene methylation predicted response to alkylating agents in glioma (Esteller et al., N Engl J Med 2000).
We have shown the relationship of methylation of MGMT with the response to alkylating agents in lymphoma (Esteller et al., J Natl Cancer Inst, 2002); of WRN with the response to irinotecan (Agrelo et al., Proc Natl Acad Sci USA, 2006); of BRCA1 with the response to PARP inhibitors (Veeck et al., J Natl Cancer Institute, 2010) and of DERL3 with the response to glycolysis inhibitors (Lopez-Serra et al., Nature Communications, 2014). Methylation of SRBC (Moutinho et al. J Natl Cancer Institute, 2014) and SLFN11 (Nogales et al., Oncotarget 2015) have also been identified as resistance markers for platinum derivatives in human tumours and the regulator of EGFR TBC1D16 has been identified as a sensitizer for therapies with BRAF and MEK inhibitors (Vizoso et al., Nature Medicine 2015). Epigenetic loss of SVIP is also related to the response to GLUT1 inhibitors (Llinas-Arias et al. JCI Insight 2019). From a multiomics standpoint, we have contributed to the characterization of drug sensitivity in 1,000 cancer cell lines (Iorio et al., Cell 2016), unveiled the reasons for those patients described as “exceptional responders” (Wheeler et al., Cancer Cell 2021), deciphered the molecular substrates behind look-alike humans (Joshi et al., Cell Reports 2022), and characterized at the single-cell multiomics level the effect of a hypomethylating agent in myelodysplastic syndrome patients (Campillo-Marcos et al., Cancer Research Communications 2024).
OUR CHALLENGES
Continuing with this translational side of our work, we are also interested in the development and study of new epigenetic drugs that target DNA methylation and histone modification writers, readers and erasers and could have an anti-cancer effect (Lara et al Oncogene 2008; Zubia et al Oncogene 2009; Huertas et al., Oncogene 2012; Perez-Salvia et al., Oncotarget 2017; Perez-Salvia et al., Haematologica 2018).
Interestingly, the “repertoire” of epigenetic modifications of DNA is fairly limited, as we recently reviewed (Heyn and Esteller, Cell 2015). In sharp contrast, more than one hundred post-transcriptional modifications occur in RNA (Esteller and Pandolfi, Cancer Discovery 2017; Davalos et al., Cell 2018; Rosselló-Tortella, Ferrer and Esteller, Blood Cancer Discovery 2020; Orsolic et al., Trends in Genetics, 2023).
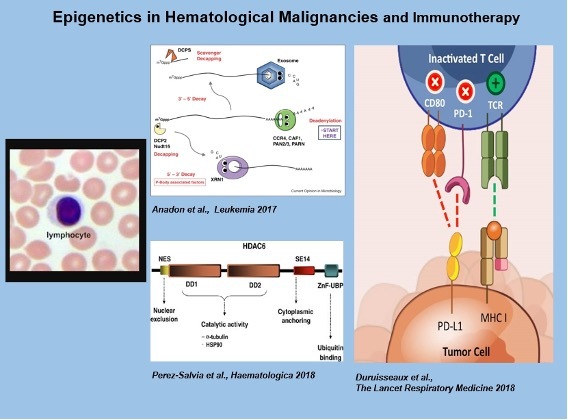
Until very recently it was almost impossible to make a good map of the epigenetic modifications of the RNA molecule, which hampered many studies in this area and prevented advances in the study of the significance of each RNA modification. However, recent methodologies now allow the study of the so-called epitranscriptome. In this field, we have shown aberrant RNA editing mediated by ADAR1 amplification in lung cancer (Anadon et al., Oncogene 2016), altered RNA decapping mediated by NUDT16 epigenetic silencing in T-ALL (Anadon et al., Leukemia 2017), RNA methylation loss in ribosomal RNA in glioma (Janin et al., Acta Neuropathol 2019), unpaired guanine modification of transfer RNA in colon cancer (Rosselló-Tortella et al., PNAS 2020), m1A defects in Hodgkin’s lymphoma (Esteve-Puig et al., Blood 2020), epigenetic disregulation of tRNAs in several tumor types (Rosselló-Tortella et al., Molecular Cancer, 2022), silencing of the 5mC RNA methyltransferase NSUN7 in liver cancer (Portiz-Barahona et al., Molecular Cancer 2023) and the contribution of m6A RNA shifts in cellular transdifferentiation (Bueno-Costa et al., Leukemia 2022). Knowledge in this area is limited and its study is the focus of intense research in the lab.
We have also a long-standing vocation for research in monogenic disorders affecting epigenetic genes (Urdinguio et al., Lancet Neurol. 2009), particularly in Rett syndrome. The disease is associated with a germline mutation in MECP2, a protein that it is attracted to methylated DNA. Over the years, we have identified the gene targets for MECP2 (Ballestar et al., EMBO J 2013; Petazzi et al. RNA Biol. 2013, Neurobiol Dis. 2014), studied the genomics of Rett syndrome in detail (Saez et al., Genet Med 2016; Lucariello et al., Hum Genet 2016) and developed pre-clinical drug studies (Szczesna et al., Neuropsychopharmacology et al., 2014; Jorge-Torres et al., Cell Reports 2018).
In a similar context, we are also curious about the epigenomic profiles of common diseases such as cardiovascular alterations (Zaina et al., Circ Cardiovasc Genet. 2014; Valencia-Morales et al., BMC Med Genomics 2015) and Alzheimer and other neurodegenerative diseases (Sanchez-Mut et al., Brain et al., 2013; Hipoccampus 2014; Transl Psychiatry. 2016; Nature Medicine, 2018).
Finally, we have a strong interest in the establishment of new epigenomic platforms to elaborate comprehensive DNA methylome maps, our lab is the pioneer in the validation of the commonly used DNA methylation microarrays such as the 450K (Sandoval et al., Epigenetics 2011), the EPIC/850K (Moran et al. Epigenomics 2016) and its new V2 version (Noguera-Castells et al. Epigenetics, 2023), plus the mouse DNA methylation microarray (García- Prieto et al., Epigenetics 2022). The use of these approaches has made several breakthroughs possible, such as: the establishment of DNA methylation signatures that are associated with early dissemination in lung cancer (Sandoval et al., JCO 2010); the diagnosis of the tumor type in Cancer of Unknown Primary (CUP) (Moran et al., Lancet Oncology 2016); the better understanding of the response to anti-PD1 immunotherapy (Duruisseaux et al., The Lancet Respiratory Medicine 2018); the obtention of the first DNA methylome of CAR-T cells with clinical value (Garcia-Prieto et al., J National Cancer Institute 2021), the prediction of COVID-19 clinical severity according to the epigenetic setting in adult (Castro de Moura et al., Lancet EBioMedicine 2021) and children (Davalos et al., Lancet EclinicalMedicine 2022) or the epigenetic scars left in the lung by SARS-CoV-2 infection (Noguera-Castells et al., CHEST 2023).
Team

Manel Esteller
Senior group leader
Senior group leader

Esteban Fernando Setién
Research Associate
Research Associate

Eva Musulén
Research Associate
Research Associate

Gerardo Ferrer
Research Associate
Research Associate

Maria Verónica Dávalos
Research Associate
Research Associate

Aleix Noguera
Postdoctoral Investigator
Postdoctoral Investigator

Alex Martinez-Sabadell
Postdoctoral Investigator
Postdoctoral Investigator

Enrique Javier Arenas
Postdoctoral Investigator
Postdoctoral Investigator

Eva Crespo
Postdoctoral Investigator
Postdoctoral Investigator

Ignacio Campillo
Postdoctoral Investigator
Postdoctoral Investigator

Ines Oršolić
Postdoctoral Investigator
Postdoctoral Investigator

Reid Elizabeth Blanchett
Postdoctoral Investigator
Postdoctoral Investigator

Aleix Franquesa
PhD Student
PhD Student

Carlos Quero
PhD Student
PhD Student

Eloy Santos
PhD Student
PhD Student
Karim Yassine
PhD Student
PhD Student

Lovro Martinovic
PhD Student
PhD Student

Tamar Levy
PhD Student
PhD Student

Victor Sanchez
PhD Student
PhD Student

Yoana Veselinova
PhD Student
PhD Student

Marta Soler
Senior Lab Technician
Senior Lab Technician
Selected Publications
Current Grants
CPP2023-010525
Ministerio de ciencia e innovación
COMBATIR NUEVAS ESTRATEGIAS COMBINATORIAS Y BIOMARCADORES DE RESPUESTA A INHIBIDORES DE HDAC6 PARA EL TRATAMIENTO DEL CÁNCER.
European academy of dermatology and venereology
SCIENCE-SS Single-cell Characterization of Epigenetic and Transcriptomic Dynamics in Sézary Syndrome Progression
HR24-00788
Fundació "la caixa"
Solve-ALD Brain Inflammation in adrenoleukodystrophy: from drivers to treatments
PMPER24/00017
Instituto de salud carlos iii
SEED-ALS Synergizing Efforts to Develop and Accelerate Breakthroughs in ALS Research
GLD24/00098
Gilead sciences s.l.
Identification of DLBCL key molecular features for CAR-T cell therapy
HR24-00788
Fundació "la caixa"
Solve-ALD Brain Inflammation in adrenoleukodystrophy: from drivers to treatments
A29372/GEACC19003CED
Fundación científica de la asociación española contra el cáncer
PREDICT-Meso PRE-malignant Drivers Combined with Target-Drug validation in Mesothelioma
OC-2023-1-26592
European commission
SENESCENCE2030 Targeting Cell Senescence to Prevent Age-Related Diseases
DJCLS 03 R/2024
Deutsche josé carreras leukämie stiftung
scEpiCART Refinement of Epigenetic Factors to Improve CAR T Cell Therapeutic Efficacy
AP183262023
Fundación mutua madrileña
Identificación de determinantes de respuesta a quimioinmunoterapia en biopsia líquida, en pacientes con cáncer microcítico de pulmón.
RETOS245779LLOV
Ministerio de ciencia e innovación
ASPIRE Neoadjuvant-adjuvant immunotherapy and cancer vaccines to improve survival in hepatocellular carcinoma
CB16/12/00312
Instituto de salud carlos iii
CIBERONC Centro de Investigación Biomédica en Red Cáncer
Cll global research foundation
Gene Expression Regulation Study Mediated by the RNA-Binding Protein Musashi2 and the RNA Methyladenosine Transferase 3, as Therapeutic Targets in Chronic Lymphocytic Leukemia
HR22-00732
Fundació "la caixa"
MyoClonal Somatic mutations and clonal hematopoiesis as predictors and drivers of heart failure progression
101136622
European commission
THRIVE TUMOUR-HOST INTERACTIONS IN LIVER CANCER OF CHILDHOOD AND ADULTS
FJC2020-044658-I
Ministerio de ciencia e innovación
Single cell analysis of clonal heterogeneity in myelodysplastic syndromes treated with azacitidine
FPU22/01655
Ministerio de universidades
Estudios de epigenética del cáncer a nivel de célula única
TALEN258406BLAN
Fundación científica de la asociación española contra el cáncer
T-REX Transcriptional Role and Epigenetics of the X chromosome in leukemias
101126676
European commission
IRB-TARGET IRB Barcelona International PhD programme: on TARGET for high-impact biomedicine
2024 FI-1 00954
Agència de gestió d'ajuts universitaris i de recerca
Overcoming Cancer Immunotherapy Resistance
PRE2022-105015
Ministerio de ciencia e innovación
USO DE APROXIMACIONES DE CELULA UNICA PARA DESCIFRAR LA EPIGENOMICA DEL CANCER Y LAS EPIDROGAS